Breath Analysis: A Potential Ally for Cystic Fibrosis
Published on: 1 Mar 2024
Cystic fibrosis is a life-shortening respiratory disease affecting approximately 10,000 people in the UK and is the most common genetic disease affecting the Caucasian population1. It is caused by an autosomal recessive mutation affecting the gene that encodes the cystic fibrosis transmembrane conductance regulator protein (CFTR), a membrane channel protein, resulting in the defective process of mucociliary clearance in the airways (Fig 1). Subsequent accumulation of viscous secretions creates an environment prone to chronic infection from pathogens, leading to inflammation, impairing respiratory function, and eventually causing death from respiratory failure2.
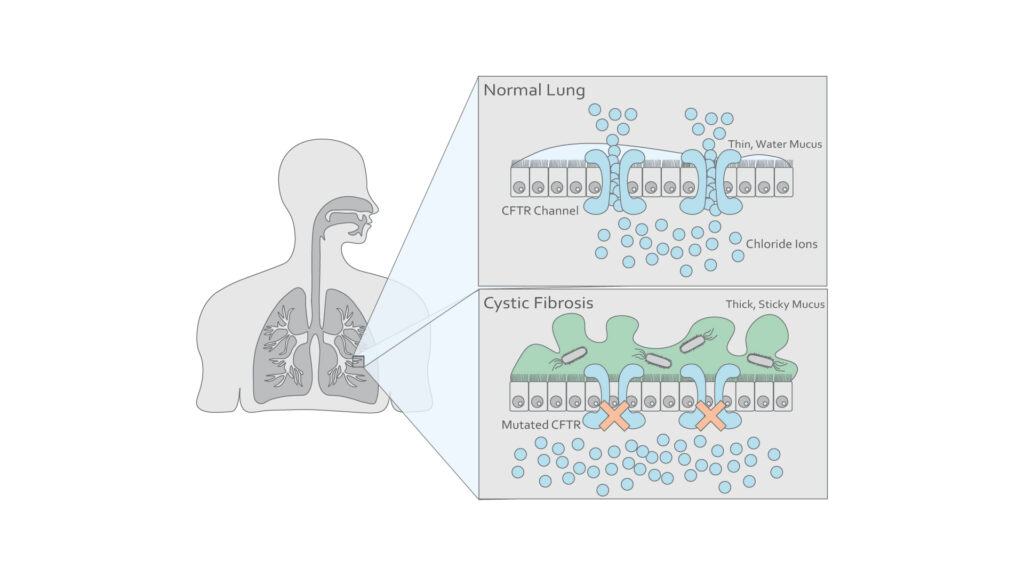
Figure 1: Illustration of epithelial cells in a healthy lung and one affected by cystic fibrosis. (Adapted from Chirico V. et al3)
The compromised airways of individuals with cystic fibrosis provides a perfect environment for Aspergillus fumigatus to colonize. This can lead to allergic bronchopulmonary aspergillosis (ABPA), a particularly severe disease affecting 8-10% of patients4. Diagnosis of ABPA is difficult as many of the symptoms overlap with those of cystic fibrosis5 and often goes undiagnosed, leading to delays in treatment which results in a poorer patient prognosis6.
Aspergillus fumigatus produces unique secondary metabolites that are volatile and therefore detectable in breath samples7. Breath analysis could therefore offer a non-invasive diagnostic solution to detect Aspergillus fumigatus in cystic fibrosis patients.
Understanding Cystic Fibrosis
There have been approximately 2000 mutations identified affecting the CFTR gene in patients with cystic fibrosis8. These mutations result in a failure of CFTR-mediated chloride and bicarbonate– transport, necessary for the unfolding and release of mucins8. Mucins are high-molecular-weight glycoproteins responsible for the viscosity of mucus. The resulting change in ionic concentrations leads to an alteration to the biophysical properties of mucus, leading to reduced innate immune defenses in affected tissues and a greater propensity to infection9.
Recent advances have resulted in the drug Trikafta being incorporated in treatment plans. Trikafta acts to modulate the defects caused by the mutated CFTR gene and is a combination of three different medications. Ivacaftor and Elexacaftor increase chloride transport through binding to separate locations on the CFTR protein whilst Tezacaftor assists in the correct folding of the CFTR protein. With Trikafta, the function of mutant CFTR proteins is greatly increased, reducing symptoms10.
Established monitoring techniques tend to rely on sputum as a source of biomarkers but Trikafta reduces sputum production in such a way as to make sputum sample collection impossible in a majority of patients11. Therefore, non-invasive monitoring using breath analysis could provide an attractive alternative source of biomarkers following treatment.
Dr. Kavita Jeerage from the National Institute of Standards and Technology (NIST) recently presented at the Breath Biopsy Conference 2024, on ‘Potential Uses of Breath Surrogates for the Development and Deployment of New Infectious Disease Biomarkers’. You can watch Dr. Jeerage’s talk on-demand, as well as the rest of the talks from the conference here.
Aspergillus infection in individuals
In healthy individuals, Aspergillus fumigatus spores that enter the lungs are killed off by the mucociliary elevator and alveolar macrophages. However, in cystic fibrosis patients the more viscous mucus in the alveoli traps the spores, reducing the ability of the immune system to act upon them. Abnormal CFTR expression also impairs the generation of antimicrobials in neutrophils contributing to colonization of the airways4.
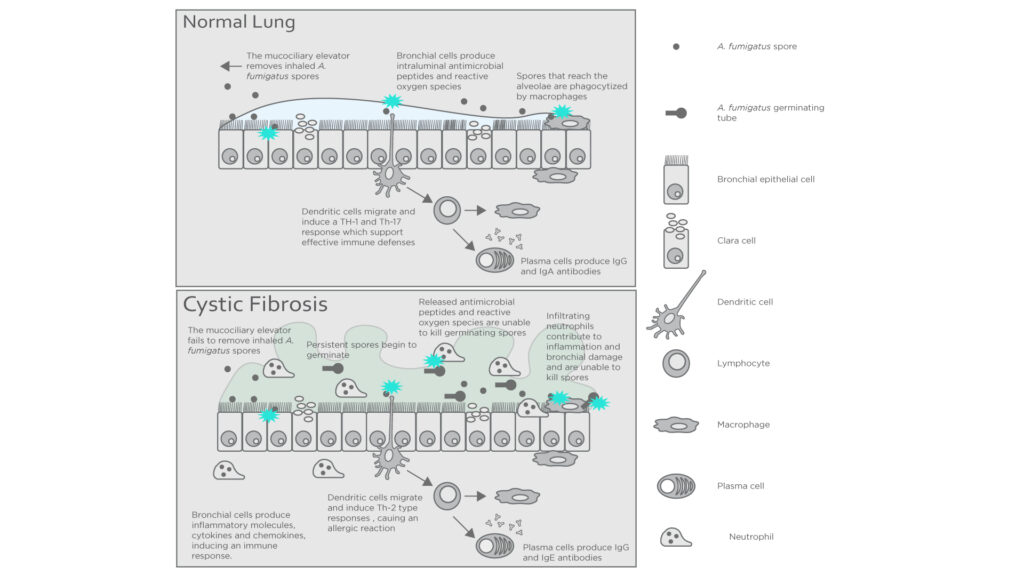
Figure 2: Interactions between the immune system and A. fumigatus in a healthy host and one with cystic fibrosis. Adapted from Carsin A. et al4.
ABPA presents very similarly to cystic fibrosis including symptoms like coughing, bronchospasm, intercurrent infections, and radiological abnormalities which complicate the monitoring of treatment progression. Furthermore, treatment often includes using a combination of corticosteroids and systemic antifungals, making accurate and precise monitoring incredibly important. Excessive treatment with corticosteroids can inhibit the immune system whilst antifungals may cause dysbiosis of the patient’s microbiome4. Thus, there is a need for a means of longitudinal testing throughout the treatment process. The volatile nature of many of the secondary metabolites produced by Aspergillus fumigatus means that breath analysis could offer a convenient and non-invasive method for this purpose.
Breath Analysis: Differentiating levels of volatile compounds between patients and controls
Detecting emerging pulmonary diseases, especially in young children with cystic fibrosis poses challenges due to invasive techniques and complications associated with late detection. Airway inflammation, a central process in chronic respiratory diseases like cystic fibrosis, asthma, COPD, and lung cancer, offers potential breath-based biomarkers for ongoing monitoring.
In chronic lung diseases, inflammation exhibits diverse mechanisms and triggers. Elevated reactive oxygen species, a common sign inflammation, leads to lipid peroxidation, generating detectable VOCs in breath. Robroeks et al. demonstrated the potential of VOCs to not only differentiate cystic fibrosis patients from controls but also distinguish patients with and without Pseudomonas colonization12. A model using 22 VOCs accurately differentiated CF children from controls, and a separate model with 14 VOCs distinguished CF subjects with and without Pseudomonas colonization with 100% accuracy. Early differentiation is crucial, especially given P. aeruginosa infection is associated with increased morbidity and mortality in CF patients.
The Breath Biopsy® Collection Station offers a non-invasive and highly reproducible method for in vivo sample collection. If you would like to find out more about using Breath Biopsy to tackle respiratory diseases such as cystic fibrosis, you can take a look at our respiratory hub page, or get in touch to explore incorporating our technology into your biomarker discovery workflow.
References:
- Keogh RH, Szczesniak R, Taylor-Robinson D, Bilton D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J Cyst Fibros. 2018 Mar;17(2):218-227. doi: 10.1016/j.jcf.2017.11.019
- Lahiri T, Sullivan JS. Recent advances in the early treatment of cystic fibrosis: Bridging the gap to highly effective modulator therapy. Pediatr Pulmonol. 2022 Feb;57 Suppl 1:S60-S74. doi: 10.1002/ppul.25660.
- Chirico, V., et al., Acute pulmonary exacerbation and lung function decline in patients with cystic fibrosis: high-mobility group box 1 (HMGB1) between inflammation and infection. Clin Microbiol Infect, 2015. 21(4): p. 368 e1-9. doi: 10.1016/j.cmi.2014.11.004
- Carsin A, Romain T, Ranque S, Reynaud-Gaubert M, Dubus JC, Mège JL, Vitte J. Aspergillus fumigatus in cystic fibrosis: An update on immune interactions and molecular diagnostics in allergic bronchopulmonary aspergillosis. Allergy. 2017 Nov;72(11):1632-1642. doi: 10.1111/all.13204
- Janahi IA, Rehman A, Al-Naimi AR. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann Thorac Med. 2017 Apr-Jun;12(2):74-82. doi: 10.4103/atm.ATM_231_16
- Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Diagnosing Allergic Bronchopulmonary Aspergillosis: A Review. Cureus. 2019 Apr 27;11(4):e4550. doi: 10.7759/cureus.4550
- Sophia Koo, Horatio R. Thomas, S. David Daniels, Robert C. Lynch, Sean M. Fortier, Margaret M. Shea, Preshious Rearden, James C. Comolli, Lindsey R. Baden, Francisco M. Marty, A Breath Fungal Secondary Metabolite Signature to Diagnose Invasive Aspergillosis, Clinical Infectious Diseases, Volume 59, Issue 12, 15 December 2014, Pages 1733–1740, doi.org 10.1093/cid/ciu725
- Milla CE, Moss RB. Recent advances in cystic fibrosis. Curr Opin Pediatr. 2015 Jun;27(3):317-24. doi: 10.1097/MOP.0000000000000226
- Morrison CB, Markovetz MR, Ehre C. Mucus, mucins, and cystic fibrosis. Pediatr Pulmonol. 2019 Nov;54 Suppl 3(Suppl 3):S84-S96. doi: 10.1002/ppul.24530
- Dawood SN, Rabih AM, Niaj A, Raman A, Uprety M, Calero MJ, Villanueva MRB, Joshaghani N, Villa N, Badla O, Goit R, Saddik SE, Mohammed L. Newly Discovered Cutting-Edge Triple Combination Cystic Fibrosis Therapy: A Systematic Review. Cureus. 2022 Sep 20;14(9):e29359. doi: 10.7759/cureus.29359
- Aridgides DS, Mellinger DL, Gwilt LL, Hampton TH, Mould DL, Hogan DA, Ashare A. Comparative effects of CFTR modulators on phagocytic, metabolic and inflammatory profiles of CF and nonCF macrophages. Sci Rep. 2023 Jul 25;13(1):11995. doi: 10.1038/s41598-023-38300-9
- Robroeks, C., van Berkel, J., Dallinga, J. et al. Metabolomics of Volatile Organic Compounds in Cystic Fibrosis Patients and Controls. Pediatr Res 68, 75–80 (2010). doi: 10.1203/PDR.0b013e3181df4ea0